CONTENTS
Definition and Structure
Classification of viruses
Replication of viruses
DNA virus replication
RNA virus replication
Plus-stranded RNA viruses
Replication of polio virus
Negative-stranded RNA viruses
Replication of Vesicular stomatatis virus (VSV)
Double-stranded RNA viruses
SV40 replication
Production of viral mRNA’s and proteins
Retroviruses & their replication
Replication of retrovirus (HIV)
Oncogenic viruses
Acute virus infections
Hepatitis A virus
Hepatitis B virus
Acquired Immunodeficiency Syndrome (AIDS)
Influenza virus
Vaccines in prevention of viral infections
Definition
Viruses exist wherever life is found. Without a host cell, viruses cannot carry out their life sustaining functions or reproduce. They are obligate intracellular parasites which infect all major groups of organisms: vertebrates, invertebrates, plants, fungi, bacteria but some viruses have a broader host range than others; however, none can cross the eukaryotic/prokaryotic boundary.
Virus structure
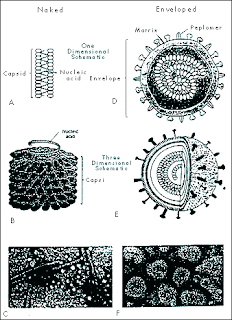
Viruses range in size from less than 100 nanometers in diameter to several hundred nanometers in length. All viruses contain a nucleic acid genome (RNA or DNA) and a protective protein coat, the capsid. The nucleocapsid may have icosahedral, helical or complex symmetry. Viruses may or may not have an envelope. Enveloped viruses obtain their envelope by budding through a host cell membrane. In some cases, the virus buds through the plasma membrane but in other cases the envelope may be derived from other membranes such as those of the Golgi body or the nucleus. Some viruses bud throughspecialized parts of the plasma membrane of the host cell; for example, Ebola virus associateswith lipid rafts that are rich in sphingomyelin, cholesterol. Poxviruses are exceptional in that they wrap themselves in host cell membranes using a mechanism that is different from the usual budding process used by other viruses.
Classification of viruses
Viruses can be classified on different basis (Proposed by David Baltimore) :
Viruses can be classified on different basis (Proposed by David Baltimore) :
- Geometry,
- Envelopes (enveloped or non-enveloped viruses),
- Identity of the host organism they can infect (bacteriophage, animal viruses, plant viruses),
- Mode of transmission,
- Structure and composition of virus particle or
- Disease they cause.
- Nucleic acid the virus contains and
- Mode of expression.
1. Virus morphology
Viruses, as viewed through the electron microscope, come in a variety of shapes (i.e. morphologies) that may be divided into (Fig.1a):
i. Helical viruses: Helical viruses are nonenveloped with capsomeres, which arearranged helically around the virus genome. Example: Tobacco Mosaic Virus

ii. Polyhedral viruses (or icosahedral): Polyhedral viruses are nonenveloped viruses whose capsids form geometric shapes with flat sides (i.e., faces) and edges. Example: Reovirus, Adenovirus, and Picornavirus.


iii. Enveloped viruses: Enveloped helical viruses are enveloped viruses whose envelope surrounds a capsid with helical virus morphology. Example:Paramyxovirus


iv. Enveloped polyhedral viruses: These are enveloped viruses whose envelope surrounds a capsid with polyhedral virus morphology. Example: Herpesvirus andTogavirus
v. Complex viruses: The morphology of complex viruses consists of complex combinations of structures that may or may not be completely consistent between viruses of the same species. Example: Tailed bacteriophages are complex viruses.
2. Virus genome
Classification by genome type :
DNA viruses
DNA viruses
· Group I - dsDNA viruses (double stranded DNA). Example: Simian virus 40 (SV 40)
· Group II - ssDNA viruses (single stranded DNA). Example: Parvovirus.
RNA viruses
RNA viruses
· Group III - dsRNA viruses (double stranded RNA). Example: Rotavirus
· Group IV - (+)ssRNA viruses (positive single stranded RNA or mRNA like).
Examples: Coronavirus, Hepatitis E virus, Hepatitis A virus, Polio virus, Rubella virus
Examples: Coronavirus, Hepatitis E virus, Hepatitis A virus, Polio virus, Rubella virus
· Group V - (-)ssRNA viruses (negative single-stranded RNA). Examples: Mumps virus, Measles virus, Rabies virus, Influenza virus.
DNA and RNA Reverse Transcribing viruses:
DNA and RNA Reverse Transcribing viruses:
· Group VI - ssRNA-RT viruses (single stranded RNA). Example: Retrovirus.
· Group VII - dsDNA-RT viruses (double stranded DNA). Example: Hepatitis Bvirus.
Replication of viruses
1. Adsorption
The first step in infection of a cell is attachment to the cell surface. Attachment is via ionic interactions, which are temperature-independent. The viral attachment protein recognizes specific receptors, which may be protein, carbohydrate or lipid, on the outside of the cell.
2. Penetration
Some enveloped viruses fuse directly with the plasma membrane. Thus, the internal components of the virion are immediately delivered to the cytoplasm of the cell (e.g. Sendai virus) (Fig.) while some require an acid pH for fusion to occur and are unable to fuse directly with the plasma membrane. These viruses are taken up by invagination of the membrane into endosomes. As the endosomes become acidified, the latent fusion activity of the virus proteins becomes activated by the fall in pH and the virion membrane fuses with the endosome membrane. This results in delivery of the internal components of the virus to the cytoplasm of the cell (e.g. Influenza virus). Non-enveloped viruses may cross the plasma membrane directly or may be taken up into endosomes. They then cross (or destroy) the endosomal membrane.
3. Uncoating
Nucleic acid has to be sufficiently uncoated so that virus replication can begin at this stage. When the nucleic acid is uncoated, infectious virus particles cannot be recovered from the cell - this is the start of the eclipse phase which lasts until new infectious virions are made.
Assembly/maturation
Assembly/maturation
New virus particles are assembled. There may be a maturation step that follows the initial assembly process.
5. Release
Virus may be released due to cell lysis, or, if enveloped, may bud from the cell. Budding viruses do not necessarily kill the cell. Thus, some budding viruses may be able to set up persistent infections. Not all released viral particles are infectious. The ratio of non-infectious to infectious particles varies with the virus and the growth conditions.
DNA virus replication
The virus needs to make mRNAs that can be translated into protein by the host cell translation machinery. The virus needs to replicate its genome. Host enzymes for mRNA synthesis and DNA replication are nuclear (except for those in mitochondria) and so, if a virus is to avail itself of these enzymes, it needs to enter the nucleus. Example: Adenovirus, are icosahedral, non-enveloped viruses, 70nm in diameter. The genome is linear double stranded DNA, associated with virally coded, basic proteins in virion
Lytic cycle
Adsorption and penetration
Adenoviruses usually infect epithelial cells. They bind to a cell surface receptor and the virus is engulfed by endocytosis. The virus appears to be able to lyze endosomes. Uncoating occurs in steps. DNA is released into the nucleus (probably at a nuclear pore).
Early phase
Early transcription: Adenovirus uses host cell RNA polymerase and early mRNAs are transcribed from scattered regions of both strands. Multiple promoters result in more flexible control. mRNAs are processed by host cell capping, methylation, polyadenylation and (sometimes) splicing enzyme systems, they are then exported to the cytoplasm and translated.
The early proteins include those which:
The early proteins include those which:
• are needed for transcription (E1A protein is needed for transcription of the other early genes; as a result these other genes are sometimes referred to as "delayed early" genes and E1A is referred to as an "immediate early" gene).
• are needed for adenovirus DNA synthesis (includes DNA polymerase).
• alter expression of host cell genes. This includes genes whose products interfere with the host anti-viral response and/or interfere with cell cycle regulation.
Late phase
DNA replication: Adenovirus encodes its own DNA polymerase (which is one of the early proteins). The DNA is replicated by a strand displacement mechanism. There are no Okazaki fragments, both strands are synthesized in a continuous fashion. DNA polymerases cannot initiate synthesis de novo, they need a primer. In the case of adenovirus, the virally coded terminal protein (TP) acts as a primer. It is thus found covalently linked to the 5' end of all adenovirus DNA strands.
Late transcription:
The way in which late transcription is switched on is not well understood. Late mRNAs code predominantly for structural proteins and there is one major late promoter. The primary transcript is processed to generate various monocistronic mRNAs. There are two types of cleavage of primary transcript: (i) to generate various 3' ends which are then polyadenylated. (ii) for intron removal. It is not understood how this process is controlled such that the correct amounts of each mRNA are made. It seems that the virus makes more mRNAs and proteins than are needed for virion assembly, so precise control may not be necessary.
The way in which late transcription is switched on is not well understood. Late mRNAs code predominantly for structural proteins and there is one major late promoter. The primary transcript is processed to generate various monocistronic mRNAs. There are two types of cleavage of primary transcript: (i) to generate various 3' ends which are then polyadenylated. (ii) for intron removal. It is not understood how this process is controlled such that the correct amounts of each mRNA are made. It seems that the virus makes more mRNAs and proteins than are needed for virion assembly, so precise control may not be necessary.
Assembly
Assembly of adenovirus particles occurs in the nucleus. DNA enters the particles after immature capsids are formed. The capsids then undergo a maturation process, after which the cells lyse and virions leak out. More structural proteins are made than are needed and excess structural proteins accumulate in the nucleus where they form inclusion bodies.
RNA virus replication
RNA viruses that do not have a DNA phase
Viruses that replicate via RNA intermediates need an RNA-dependent RNA-polymerase to replicate their RNA, but animal cells do not seem to possess a suitable enzyme. Therefore, this type of animal RNA virus needs to code for an RNA-dependent RNA polymerase. No viral proteins can be made until viral messenger RNA is available. Thus, the nature of the RNA in the virion affects the strategy of the virus.
i. Plus-stranded RNA viruses
In these viruses, the virion (genomic) RNA is the same sense as mRNA and so functions as mRNA. This mRNA can be translated immediately upon infection of the host cell. Examples: Poliovirus (Picornavirus), Togaviruses, Flaviviruses.
Replication of polio virus
I. The Poliovirus Receptor (PVR)
Polio's first interaction with a host cell consists of binding to a specific cell surface protein, the poliovirus receptor (PVR). This protein, whose natural function is not known, is a member of a family of proteins called the immunoglobulin superfamily, the defining feature of which is a "loop" in the protein structure called the Ig domain. PVR has three Ig loops (which are outside the cell), numbered 1-3 starting with the loop farthest from the cell surface. The protein extends through the cell membrane, with a short stretch of amino acids (protein sequence) inside the cell as well . Polio virus appears to bind to its receptor on loop 1. This initial binding is followed by conformational changes in the virus's capsid, which are believed to prepare it for uncoating. The receptor is taken into the cell by the process of endocytosis, which is most likely involved in PVR's natural function. In other words, the virus has evolved to take advantage of a naturally occurring protein on the cell surface in order to gain entry and initiate an infection. This is a common tactic of many animal and plant viruses. The poliovirus receptor is expressed in many human tissue types, apparently including some tissues, such as kidney, which are not normal sites of poliovirus replication in the host. The tendency of a virus to replicate only in particular tissue types is called "tissue tropism," and is an active area of study for researchers working on many types of viruses. Polio virus ordinarily infects cells in the lining of the intestine and can migrate to nerve tissue, where it causes the characteristic pathology of paralytic poliomyelitis.
II. Uncoating
After binding to its receptor, poliovirus must get its genetic material into the cell's cytoplasm, where translation and replication will occur. In this respect, the viral capsid is something of a paradox, since it must be stable to harsh conditions in the environment (including the low pH of the host's stomach), but must be able to release its contents (the viral genome) easily and quickly when stimulated with the proper signal. At physiological temperatures, the virus can undergo a major structural change, called alteration, after binding to the receptor. The altered particle is easy to distinguish from the native virion, but it is unclear how - or even if - this altered stage leads to productive uncoating of the virus genome. For every 200 or so virus particles that encounter a cell, only one will successfully enter and replicate, so research in this area is often confounded by the rarity of successful entry. There are two major models for poliovirus entry. In one, the virion, after binding to PVR, initiates entry directly from the cell surface, injecting its genome into the cell's cytoplasm. In the other model, the virus particle must be taken into the cell by a process called receptor-mediated endocytosis, a mechanism routinely employed by cells to take in food and signal proteins. According to this model, the virus then uncoats inside a compartment that forms in the cell, and the genome is released into the cytoplasm. There is little experimental data to support either model, so both are considered reasonable possibilities.
III. Protein Synthesis
In contrast to the human cells it infects, which have a genome made of deoxyribonucleic acid (DNA), the poliovirus genome is made of ribonucleic acid (RNA). In a cell, RNA is used as a "messenger" to carry genetic information from the nucleus into the cytoplasm, where it is translated into proteins that are the building blocks of the cell . Poliovirus skips the DNA step and simply carries a single RNA molecule inside its protective capsid. This RNA is "message sense," meaning that it can be translated directly into proteins in the cell's cytoplasm. The entire poliovirus RNA molecule is translated into a single long "polyprotein." This large protein then cleaves itself into subsections and finally into the separate proteins involved in replication and packaging, including the virus capsid proteins. Some of the viral proteins also act to shut down the translation of the host cell's messenger RNAs while still permitting the viral RNA to be translated, making the cell a more efficient virus factory.

IV. Protein Processing
The product of translation is the long viral polyprotein which contains all of the virus's proteins strung together into a single molecule. Some of these proteins are proteases, or enzymes, which cut other proteins. In a series of cleavages, the proteases break down the polyprotein into its component parts , which then operate as separate gene products. Since the proteases are contained within the polyprotein initially, one of their most important functions is to cleave themselves out of the larger structure, freeing them to do the rest of their work. In addition to its role in cutting up the polyprotein, one of the proteases is involved in shutting off most of the host cell's own protein synthesis. The protease does this by cleaving a component of the cell's translational machinery which is required for normal protein synthesis, but which the viral RNA does not need. Shutting down the host's RNA translation serves a dual function for the virus: first, it frees up more ribosomes to translate the viral genomes, and second, it insures that the cell will die and break down, releasing the progeny virus particles after they have been assembled.
V. RNA Replication
RNA viruses have a unique difficulty when it comes to replication, as the cell does not have the necessary machinery to reproduce an RNA molecule (the cell replicates DNA, which is transcribed to produce RNA, and RNA is translated to produce proteins). This means that the virus must carry its own RNA replication proteins or have a mechanism for producing them once inside the cell. For polio, the replication functions are carried out by a viral RNA directed RNA polymerase. This means that it reads an RNA template and produces a new RNA molecule of the opposite polarity. Because RNA is single-stranded, the first round of replication produces a single antisense, or complementary, molecule. This antisense template is then used to produce a positive-sense copy of the original genome. As these new genomes accumulate, they can also act as additional messages for the cell's translation machinery, leading to higher levels of viral protein production.
VI. Packaging and Release
After the virus has translated its RNA to produce the necessary proteins and replicated its genome, it needs to package the newly synthesized RNA molecules inside capsids, or protein shells. A complete virus consists of the RNA packaged inside the capsid, which will be released from the cell for the next round of infection. The capsid proteins self-assemble into an immature capsid, a structure which contains all of the necessary proteins, but which has not finished cleaving them into their final form. The viral RNA enters the incomplete capsid and is secured inside when the viral proteases make the final cleavages. The processes, which guide the RNA to the capsid, are still poorly understood. Once the genomes have been packaged into mature virions, the virus particles await the cell's lysis (bursting), when they will be released to infect neighboring cells, starting the cycle over again.
ii. Negative-stranded RNA viruses
The virion RNA is negative sense (complementary to mRNA) and must therefore be copied into the complementary plus-sense mRNA before proteins can be made. Thus, besides needing to code for an RNA-dependent RNA-polymerase, these viruses also need to package it in the virion so that they can make mRNAs upon infecting the cell. Examples: Influenza virus (orthomyxovirus), Measles virus, Mumps virus (Paramyxoviruses), Rabies virus (Rhabdovirus), VSV virus.
Replication of Vesicular stomatatis virus (VSV)
This virus causes epidemic but self-limiting vesicular disease of cattle. Also infects swine, horses, humans and even insects (very broad host range). In humans, it causes a mild flu-like illness that's fairly common in lab workers. In keeping with its broad host range, the VSV receptor is not a protein (prolonged trypsinization of cultured cells doesn't block infection). It may be phosphatidyl serine. The morphology and structure of VSV is similar to that of rabies virus. The particles are bullet-shaped and are composed of two major structures a nucleocapsid or ribonucleoprotein (RNP) core and a lipoprotein envelope, which surrounds that core.
Viral RNP core
The nucleocapsid or RNP core is the infectious component of VSV and all other rhadbo viruses. This core includes the viral genomic RNA, which is tightly associated with the highly abundant nucleocapsid protein (N). The RNP core also contains less abundant proteins --the phosphoprotein (P), and the viral RNA polymerase (L) (Fig. 5). N protein: The function of the N protein appears to be: (i) to promote RNA encapsidation or packaging and (ii) to allow genome replication, by favouring anti-termination of transcription (i.e. by allowing the viral polymerase to read-through the stop/start signals located between the viral genes). L protein: This is the viral RNA-directed RNA polymerase. It is not active on its own, however, since P protein is needed for catalytic activity.

Viral envelope
The major components of the VSV envelope are: (i) the membrane-anchored viral glycoprotein (G) and (ii) the matrix protein (M) (Fig.). Roughly equivalent amounts of the two protein are found in each virion (approx. 1500 molecules per virion). G protein: Theglycoprotein, G, forms trimeric spikes on the surface of the viral particle and it forms both the major antigenic determinant on the virus, as well as the major receptor-binding molecule on the virus. G protein undergoes a conformational shift at mildly acidic pH (< 6.0), which stabilizes the trimer and exposes a hydrophobic domain that can insert into cellular membranes and allow membrane fusion to occur. Thus, VSV fusion is activated in the endocytic vesicle, in response to acidic pH.
Viral gene expression
After entry into its host cell, and uncoating of the RNP core, VSV begins to express its genes. Since, the viral genome is of negative sense (i.e., of opposite polarity to mRNA), the very first step is transcription of viral mRNAs. Viral transcription begins at the 3' end of the viral genome, at a single promoter element, and proceeds sequentially across the genome. It is generally believed that the individual gene-unit-length mRNAs are reproduced by a stop-start transcription mechanism. One result of this is that the transcriptase pauses and transcription is attenuated about 30% at each gene junction. This in turn produces a gradient of mRNA production, such that N>P>M>G>L.
Stop/start transcription is achieved by the presence of transcriptional signals at gene boundaries. There is a 5'-initiation signal, as well as 3'- polyA and termination signals, which are ordered: [polyAsignal/terminator]--[intergenic region]--[initiator].
Viral RNA replication
Unlike viral mRNA transcription, viral RNA replication requires the virus to form a single complete copy of its genome. The decision to replicate the viral genome must therefore be made when the first intergenic region is encountered (this is located between the region that encodes the short untranslated leader RNA and the gene encoding the N-protein). This intergenic region must be read-through in order for viral RNA replication to occur. Interestingly, viral RNA replication requires active translation (this was proved experimentally, since viral RNA replication, but not viral mRNA synthesis, was blocked by inhibition of protein synthesis using cycloheximide). This observation is consistent with a model in which newly formed viral N protein selectively binds to the viral leader RNA. By doing so, N protein prevents the recognition of transcriptional termination signals. Thus, the switch from mRNA synthesis to RNA replication is regulated principally by the anti termination activity of the N protein.
iii. Double-stranded RNA viruses
The virion (genomic) RNA is double stranded and so cannot function as mRNA; thus these viruses also need to package an RNA polymerase to make their mRNA after infection of the host cell. Example: Rotaviruses (belong to reovirus family).
SV40 replication
SV40 belongs to polyoma virus family. It is small (~40nm diameter), icosahedral, nonenveloped virus that replicates in the nucleus. Depending on the host cell, they can either transform the cell or replicate the virus and lyze the cell. SV40 infects primate cells, forcing its way inside and releasing its DNA circle. Once inside, it has two jobs: to replicate its DNA and to package it inside new viral capsids. Amazingly, SV40 only needs one protein, the Tantigen, to control both of these processes. The circular SV40 genome is found in the cell as a "mini-chromosome" wound into a handful of nucleosomes. It only has enough space to encode a few functions, since it all has to fit inside the tiny capsid. It has a regulatory region, that controls the entire lifecycle of the virus. It also encodes several proteins: the T-antigen (and a spliced version of it called the t-antigen) and three capsid proteins, VP1, VP2 and VP3.
Lytic cycle: attachment, penetration and uncoating
Viral capsid proteins interact with cell surface receptors and penetration is probably via endocytosis. Virions are transported to the nucleus and uncoated. DNA (and associated histones) enters nucleus, probably through a nuclear pore.
Production of viral mRNA’s and proteins
Gene expression is divided into early and late phases. Early genes encode enzymes and regulatory proteins needed to start viral replication processes. Late genes encode structural proteins, proteins needed for assembly of the mature virus.
Early phase of the Lytic cycle
Early gene expression (Fig.6a): The early promoter is recognized by host RNA polymerase II (SV40 contains a strong enhancer). Post transcriptional RNA modification (capping, methylation, polyadenylation, splicing, etc.) is carried out by host enzymes. The early transcript (primary transcript) is made and then undergoes alternative processing, resulting in the mRNAs for the small T and large T antigens (these proteins have common amino-termini but different carboxy-termini). The mRNAs are translated in the cytoplasm.
Late phase of the Lytic cycle
By definition the late phase starts with the onset of viral genome replication. SV40 DNA replication occurs in the nucleus. Large T antigen is needed for DNA replication. This binds to the origin of replication. SV40 virus uses the host cell DNA polymerase, which recognizes the viral origin of replication if the T antigen is present. DNA replication is bidirectional (There are two replication forks per circular DNA genome and replication involves leading/lagging strands, Okazaki fragments, DNA ligase, etc.). This process of DNA replication is very similar to that which occurs in the host cell - which is not surprising as the virus is using mainly host machinery except for the involvement of the T antigen. Host histones complex with the newly made DNA. Late mRNAs are made after DNA replication (a lot of newly made viral DNA is now available as template). Early mRNAs are still transcribed, but at a very much lower rate. T antigen is involved in controlling increased transcription from the late promoter and decreased transcription from early promoter. It also interacts with host proteins and changes the properties of the host cell, thus playing a role in cell transformation and tumour formation. VP1, 2, and 3 are made from same primary transcript which undergoes differential splicing. This results in the reading frame for VP1 being different from that for VP2 and VP3. Thus, one region of the DNA can code for two different amino acid sequences according to which reading frame is used. This is another way that viruses (and cells) can use a short stretch of DNA to code for more than one protein sequence VP1, 2 and 3 mRNAs are translated in the cytoplasm, the proteins are transported to nucleus, and capsids assemble with DNA (and cell histones) inside the capsid. Large numbers of capsids accumulate in the nucleus and form inclusion bodies. Virions are released by cell lysis.
Retroviruses & their replication
A retrovirus is a virus which has a genome consisting of two RNA molecules, which may or may not be identical. It relies on the enzyme reverse transcriptase to perform the reverse transcription of its genome from RNA into DNA, which can then be integrated into the host's genome with an integrase enzyme. The virus itself stores its nucleic acid genome and serves as a means of delivery of that genome into targeted cells, which constitute the infection. Once in the host's cell, the RNA strands undergo reverse transcription in the cytosol and are integrated into the host's genome into the germ line, their genome is passed on to a following generation. These endogenous retroviruses (vs. exogenous) now make up 8% of the human genome. Most insertions have no known function and are often referred to as "junk DNA" (Noncoding DNA). However, many endogenous retroviruses play important roles in host biology such as control of gene transcription, cell fusion during placental development, and resistance to exogenous retroviral infection. Endogenous retroviruses have also received special attention in the research of immunology- related pathologies, i.e. autoimmune
diseases such as multiple sclerosis, although endogenous retroviruses have not yet been proven to play any causal role in this class of disease.While transcription was classically thought to only occur from DNA to RNA, reverse transcriptase transcribes RNA into DNA.
The term "retro" in retrovirus refers to this reversal of the central dogma of molecular biology. Because reverse transcription lacks the usual proofreading of DNA transcription, this kind of virus mutates very often. This enables the virus to grow resistant to antiviral pharmaceuticals quickly, and impedes, for example, the development of an effective vaccine against HIV. Retrovirus genomes commonly contain these three genes, among others, that encode for proteins that can be found in the gag (group-specific antigen) codes for core and structural proteins of the virus; pol (polymerase) codes for reverse transcriptase, protease and integrase; and, env (envelope) codes for the retroviral coat proteins. Thus far, four human retroviruses have been found to attack Helper T cells.
Replication of retrovirus (HIV)
1) Adsorption
The HIV-1 envelope gp120 must attach to both a CD4 molecule (acts as a primary receptor for HIV) and a chemokine receptor (acts as a coreceptor for HIV) on the surface of such cells as macrophages and T4-helper lymphocytes in order to enter the cell. The gp120 first binds to a CD4 molecule on the plasma membrane of the host cell.
2) Penetration
The binding of a portion or domain of the HIV-1 surface glycoprotein gp120 to a CD4 molecule on the host cell induces a change in shape that brings the chemokine receptor binding domains of the gp120 into proximity with the host cell chemokine receptor (CXCR-4 or CCR-5). This, in turn, brings about a conformational change that exposes a previously buried portion of the transmembrane glycoprotein gp41 enabling the viral envelope to fuse with the host cell membrane. After fusion of the viral envelope with the host cell cytoplasmic membrane, the genome-containing protein core of the virus enters the host cell's cytoplasm.
3) Uncoating and Production of a Provirus
The single-stranded RNA genomes are released from the capsid. HIV uses the enzyme reverse transcriptase to transcribe its RNA genome into single-stranded DNA. As the DNA is being made, an RNase degrades the RNA genome. The reverse transcriptase then synthesizes a complementary DNA strand to produce a double-stranded DNA intermediate that enters the infected host cell's nucleus. An HIV enzyme called integrase is used to insert the HIV double-stranded DNA intermediate into the DNA of a host cell's chromosome. HIV is now a provirus.
4) Translation of HIV mRNA
Once synthesized, HIV mRNA goes through the nuclear pores into the rough endoplasmic reticulum to the host cell's ribosomes where it is translated into HIV structural proteins, enzymes, glycoproteins, and regulatory proteins. A 9 kilobase mRNA is formed that is used for three viral functions:
(a) Synthesis of Gag polyproteins (p55). These polyproteins will eventually be cleaved by HIV proteases to become HIV matrix proteins (MA; p17), capsid proteins (CA; p24), and nucleocapsid proteins (NC, p7).
(a) Synthesis of Gag polyproteins (p55). These polyproteins will eventually be cleaved by HIV proteases to become HIV matrix proteins (MA; p17), capsid proteins (CA; p24), and nucleocapsid proteins (NC, p7).
(b) Synthesis of Gag-Pol polyproteins (p160). These polyproteins will eventually be cleaved by HIV protease to become HIV matrix proteins (MA; p17), capsid proteins (CA; p24), proteinase molecules (protease or PR; p10), reverse transcriptase molecules (RT; p66/p51), and integrase molecules(IN;p32).
(c) During maturation, these RNA molecules also become the genomes of new HIV virions.
The 9kb mRNA can also be spliced to form a 4kb mRNA and a 2kb mRNA. The 4kb mRNA
is used to:
(a) Synthesize the Env polyproteins (gp160). These polyproteins will eventually be cleaved by proteases to become HIV envelope glycoproteins gp120 and gp41.
(b) Synthesize three regulatory proteins called vif, vpr, and vpu. The 2kb mRNA is used to synthesize three regulatory proteins known as tat, rev, and naf .
5) Maturation of Envelope Glycoproteins
The Env polyprotein (gp160) goes through the endoplasmic reticulum and is transported to the Golgi complex where it is cleaved by a protease (proteinase) and processed into the two HIV envelope glycoproteins gp41 and gp120. These are transported to the plasma membrane of the host cell where gp41 anchors the gp120 to the membrane of the infected cell.
6) Maturation and Release
Maturation either occurs in the forming bud or in the immature virion after it buds from the host cell. During maturation, HIV proteases (proteinases) will cleave the polyproteins into individual functional HIV proteins and enzymes. (a) The Gag polyproteins (p55) will be cleaved by HIV proteases to become HIV matrix proteins (MA; p17), capsid proteins (CA; p24), and nucleocapsid proteins (NC, p7 and p6). (b) The Gag-Pol polyproteins (p160) will be cleaved by HIV proteases to become HIV matrix proteins (MA; p17), capsid proteins (CA;p24), proteinase molecules (protease or PR; p10), reverse transcriptase molecules (RT; p66/p51), and integrase molecules (IN; p32). The various structural components then assemble to produce a mature HIV virion.
Oncogenic viruses
Cancers are the result of a disruption of the normal restraints on cellular proliferation. There are two basic classes of genes in which mutation can lead to loss of growth control:
(a) Those genes that are stimulatory for growth and which cause cancer when hyperactive.Mutations in these genes will be dominant.
(b) Those genes that inhibit cell growth and which cause cancer when they are turned off. Mutations in these genes will be recessive.
Viruses are involved in cancers because they can either carry a copy of one of these genes or can alter expression of the cell's copy of one of these genes.
Classes of tumour viruses
There are two classes of tumor viruses: the DNA tumor viruses and the RNA tumor viruses. These two classes have very different ways of reproducing themselves but they often have one aspect of their life cycle in common: the ability to integrate their own genome into that of the host cell. Such integration is not, however, a pre-requisite for tumour formation. If a virus takes up residence in a cell and alters the properties of that cell, the cell is said to be transformed. Transformation often includes loss of growth control, ability to invade extracellular matrix and dedifferentiation. In carcinomas, many epithelial cells undergo an epithelial-mesenchymal transformation. The region of the viral genome that can cause a tumor is called an oncogene. This foreign gene can be carried into a cell and cause it to take on new properties such as immortalization and anchorage-independent growth. The discovery of viral oncogenes in retroviruses led to the finding that they are not unique to viruses and homologous genes (called proto-oncogenes) are found in all cells. Normally, the cellular proto-oncogenes are not expressed in a quiescent cell since they are involved in growth and development; or they are expressed at low levels. However, they may become aberrantly expressed when the cell is infected by tumour viruses that do not themselves carry a viral oncogene. The discovery of cellular oncogenes led to the discovery of another class of cellular genes, the tumour repressor (suppressor) genes or anti-oncogenes.
Initially, the involvement of viral and cellular oncogenes in tumours caused by retroviruses was much more apparent than the involvement of the DNA tumour virus oncogenes but the discovery of tumour repressor genes (as a result of our knowledge of how retroviruses cause cancer) led to the elucidation of the mode of action of DNA virus oncogenes.
DNA tumour viruses
DNA tumour viruses have two life-styles: In permissive cells, all parts of the viral genome are expressed. This leads to viral replication, cell lysis and cell death. In cells non-permissive for replication, viral DNA is integrated into the cell chromosomes at random sites.
Papillomaviruses: Papilloma viruses are wart-causing viruses that also certainly cause human neoplasms and cause natural cancers in animals. Warts are usually benign but can convert to malignant carcinomas. Papilloma viruses are also found associated with human penile, uterine and cervical carcinomas. There are several types of papilloma viruses but not all are associated with cancers.
Polyomaviruses: Polyoma virus was so named because it causes a wide range of tumours in a number of animal species. It causes leukaemia’s in mice and hamsters. There are two human polyoma isolates; neither came from a tumour but they cause tumours when injected into animals. Polyoma viruses are usually lytic and when transformation occurs, after integration into host DNA, only early functions are transcribed into mRNA and expressed as a protein product. These are the tumour antigens and the expression of the genes for tumour antigens is essential for transformation of the cells. Example: SV40 large T antigen.
Adenoviruses: These viruses are highly oncogenic in animals and only a portion of the virus is integrated into the host genome. This portion codes for several T antigens. Adenoviruses cause cell transformation by the integration of early function genes into the chromosome and the expression of these DNA synthesis-controlling genes without the production of viral structural proteins.
Herpesviruses: It causes oral herpes (cold sores or fever blisters), and genital herpes (genital sores). Herpes Simplex Type 1 (HSV-1) and Herpes Simplex Type 2 (HSV-2) viruses look identical under the microscope, and either type can infect the mouth, skin or genitals. Most commonly, however, HSV-1 occurs above the waist, and HSV-2 below. Herpes is spread by direct skin-to-skin contact. There is considerable circumstantial evidence that implicates these enveloped DNA viruses in human neoplasms. They are highly tumourigenic in animals.It is notable that herpes viruses exist primarily as episomes in the cell and do not integrate into the host cell genome. By the time that tumours arise, no trace of the virus can usually be found. Herpes virus DNA is found in only a small number of herpes-transformed cells causing chromosomal breakage or other damage.
RNA tumour viruses (Retroviruses)
Retroviruses are different from DNA tumour viruses in that their genome is RNA but they are similar to many DNA tumour viruses in that the genome is integrated into host genome. Since RNA makes up the genome of the mature virus particle, it must be copied to DNA prior to integration into the host cell chromosome.
Oncogenes in retroviruses
If the virus is to transform a cell, in addition to, or instead, of part of the gag/pol/env genome, it must have sequences that alter cellular DNA synthesis and provide the other functions that are typical of a transformed cell. Thus we also find an Oncogene (onc) in the viral genome of many retroviruses that transform cells to neoplasia. The changes in the biologic function and antigenic specificity of a cell that result from integration of viral genetic sequences into the cellular genome and that confer on the infected cell certain properties of neoplasia. In retroviruses, these were first discovered as an extra gene in Rous sarcoma virus (RSV). This gene was called src (for sarcoma). src is not needed for viral replication. It is an extra gene to those (gag/pol/env) necessary for the continued reproduction of the virus. RSV has a complete gag/pol/env genome. Deletions/mutations in src abolish transformation and tumour promotion but the virus is still capable of other functions. RSV is unusual in that it has managed to retain its whole genome of gag/pol/env. In sharp contrast to RSV, many retroviruses have lost part of their genome to accommodate an oncogene. About forty oncogenes have now been identified. they are referred so by a three letter code (e.g. src, myc) often reflecting the virus from which they were first isolated. Some viruses can have more than one oncogene (e.g. erbA, erbB).
Oncovirinae: These are the tumour viruses and the first member of this group to be discovered was Rous sarcoma virus (RSV) - which causes a slow neoplasm in chickens. HTLV-1 (human T-cell lymphotropic virus): causes adult T-cell leukaemia. HTLV-1 is sexually transmitted and causes tumour in humans.
Lentivirinae: These have a long latent period; they are mainly associated with diseases of ungulates (e.g. visna virus), but HIV, which causes AIDS, belongs to this group.
Acute virus infections
A. Hepatitis A virus
Hepatitis A (HAV) is caused by a RNA virus, which is found in faeces, saliva, semen, and blood of infected people. It is transmitted primarily by the faecal, oral or sexual route, but can be passed rarely by blood transfusion or contaminated needles. It is icosahedral non-enveloped single- stranded, positive sense virus measuring approximately 28 nm in diameter. In humans, viral replication depends on hepatocyte uptake and synthesis, and assembly occurs exclusively in liver cells. Various genotypes of the virus exist; however, there appears to be only one serotype. Hepatocyte uptake involves a receptor on the plasma membrane of the cell, and viral replication is believed to occur exclusively in hepatocytes. After entry into the cell, viral RNA is uncoated, and host ribosomes bind to form polysomes. Viral proteins are synthesized, and a viral RNA polymerase copies the viral genome. Assembled virus particles are shed into the biliary tree and excreted in the faeces. Minimal cellular morphologic changes result from hepatocyte infection. The development of an immunologic response to infection is accompanied by a predominantly portal and periportal lymphocytic infiltrate and varying degree of necrosis. Person-to-person contact is the most common means of transmission and is generally limited to close contacts. The incubation period usually lasts two to six weeks, and the time to onset of symptoms may be dose related. The presence of disease manifestations and the severity of symptoms following infection directly correlate with patient age. In developing nations, the age of acquisition is usually before age 2 years; in Western societies, acquisition is most frequent in persons aged 5-17 years. In this age range, the illness is more often mild or subclinical; however, severe disease may result in complete hepatic failure.
B. Hepatitis B virus
Its genome consists of a partially double-stranded circular DNA of 3.2 kilobase pairs that encodes 4 overlapping open reading frames:
S for the surface or envelope gene encoding the pre-S1, pre-S2, and the S protein.
C for the core gene, encoding for the core nucleocapsid protein and the E antigen.
X for the X gene encoding the X protein.
P for the polymerase gene encoding a large protein promoting priming, RNA-dependent
and DNA-dependent DNA polymerase and RNase H activities.
An upstream region for the S and C genes has been found, named pre-S and pre-C, respectively. The structure of this virion is a 42-nm spherical double-shelled particle consisting of small spheres and rods, with an average width of 22 nm. The S gene encodes the viral envelope. There are mainly five antigenic determinants: A, common to all HBsAg and D, Y, W, and R, which are epidemiologically important. The core antigen, HBcAg, is the protein that encloses the viral DNA. It also can be expressed on the surface of the hepatocytes, initiating a cellular immune response. The E antigen, HBeAg, comes from the core gene and is a marker of active viral replication. Usually, HBeAg can be detected in patients with circulating serum HBV DNA. The best indication of active viral replication is the presence of HBV DNA in the serum. Hybridization or more sensitive polymerase chain reaction (PCR) techniques are used to detect the viral genome in the serum. It is an extremely resistant strain capable of withstanding extreme temperatures and humidity. It can survive when stored for 15 years at -20°C, for 24 months at -80°C, for 6 months at room temperatures, and for 7 days at 44°C. The role of the X gene is to encode proteins that act as transcriptional transactivators aiding viral replication. Evidence strongly suggests that these transactivators may be involved in carcinogenesis. The production of antibodies against HBsAg confers protective immunity and can be detected in patients who have recovered from HBV infection or in those who have been vaccinated. Antibody to HBcAg is detected in almost every patient with previous exposure to HBV. The immunoglobulin M (IgM) subtype is indicative of acute infection or reactivation, while the immunoglobulin G (IgG) subtype is indicative of chronic infection. With this marker alone, one cannot understand the activity of the disease. Antibody to HBeAg is suggestive of a non-replicative state, and indicates that the antigen has been cleared. The pathogenesis and clinical manifestations are due to the interaction of the virus and the host immune system. The latter attacks the HBV and causes liver injury. Impaired immune reactions (eg, cytokine release, antibody production) or relatively tolerant immune status results in chronic hepatitis. In particular, a restricted T cell–mediated lymphocytic response occurs against the HBV-infected hepatocytes. The final state of the disease is cirrhosis. Patients with cirrhosis and HBV infection are likely to develop hepatocellular carcinoma (HCC). In the United States, the most common presentation is that of patients of Asian origin who acquired the disease as newborns (vertical transmission).
Four different stages have been identified in the viral life cycle.
The first stage is immune tolerance. The duration of this stage for healthy adults is approximately 2-4 weeks and represents the incubation period. For newborns, the duration of this period often is decades. Active viral replication is known to continue despite little or no elevation in the aminotransferase levels and no symptoms of illness.
In the second stage, an inflammatory reaction with a cytopathic effect occurs. HBeAg can be identified in the sera, and a decline of the levels of HBV DNA is seen. The duration of this stage for patients with acute infection is approximately 3-4 weeks (symptomatic period). For patients with chronic infection, 10 years or more may elapse before cirrhosis develops.
In the third stage, the host can target the infected hepatocytes and the HBV. Viral replication no longer occurs, and HBeAb can be detected. The HBV DNA levels are low or undetectable, and aminotransferase
In the second stage, an inflammatory reaction with a cytopathic effect occurs. HBeAg can be identified in the sera, and a decline of the levels of HBV DNA is seen. The duration of this stage for patients with acute infection is approximately 3-4 weeks (symptomatic period). For patients with chronic infection, 10 years or more may elapse before cirrhosis develops.
In the third stage, the host can target the infected hepatocytes and the HBV. Viral replication no longer occurs, and HBeAb can be detected. The HBV DNA levels are low or undetectable, and aminotransferase
levels are within the reference range. In this stage, an integration of the viral genome into the host's hepatocyte genome takes place. HBsAg is still present.
In the fourth stage, the virus cannot be detected and antibodies to various viral antigens have been produced.
Different factors have been postulated to influence the evolution of these stages, including age, sex, immunosuppression, and co-infection with other viruses. Eight different genotypes A through H representing a divergence of the viral DNA at around 8% have been identified. The prevalence of the genotypes varies in different countries. The progression of the disease seems to be more accelerated, and the response to treatment with antivirals is less favorable for patients infected by genotype C compared with those infected by genotype B.
In the fourth stage, the virus cannot be detected and antibodies to various viral antigens have been produced.
Different factors have been postulated to influence the evolution of these stages, including age, sex, immunosuppression, and co-infection with other viruses. Eight different genotypes A through H representing a divergence of the viral DNA at around 8% have been identified. The prevalence of the genotypes varies in different countries. The progression of the disease seems to be more accelerated, and the response to treatment with antivirals is less favorable for patients infected by genotype C compared with those infected by genotype B.
C. Acquired Immunodeficiency Syndrome (AIDS)
AIDS is the name given to end-stage disease caused by human immunodeficiency virus (HIV). Initial description of the human immunodeficiency virus type I (HIV-1) in 1983 and HIV-2 in 1986, these two viruses have been identified for almost 20 years as the primary cause of the acquired immunodeficiency syndrome (AIDS). The course of infection with HIV-1 in HIV-infected humans may vary dramatically, even if the primary infections arose from the same source. In some individuals, with a long-term non-progressive HIV-1 infection (i.e., lack of decline in CD4 counts, or chronic infection for at least 7 years without the development of AIDS), a defective virion was identified. Thus, infection with a defective virus, or one which has a poor capacity to replicate, may prolong the clinical course of HIV-1 infection.
The structure of HIV-1
HIV-1 is a retrovirus and belongs to the family of lentiviruses. Infections with lentiviruses typically show a chronic course of disease, a long period of clinical latency, persistent viral replication and involvement of the central nervous system. Using electron microscopy, HIV-1 and HIV-2 resemble each other strikingly. However, they differ with regard to the molecular weight of their proteins, as well as having differences in their accessory genes. Both HIV-1 and HIV-2 replicate in CD4 T cells and are regarded as pathogenic in infected persons, although the actual immune deficiency may be less severe in HIV-2-infected individuals.
HIV-1 viral particles have a diameter of 100 nm and are surrounded by a lipoprotein .
.
.
The Organization of the Viral Genome
Most replication competent retroviruses depend on three genes: gag, pol and env: gag means "group-antigen", pol represents "polymerase" and env is for "envelope". The "classical" structural scheme of a retroviral genome is: 5'LTR-gag-pol-env-LTR 3'. The LTR ("long terminal repeat") regions represent the two end parts of the viral genome, that are connected to the cellular DNA of the host cell after integration and do not encode for any viral proteins.
The gag and env genes code for the nucleocapsid and the glycoproteins of the viral membrane; the pol gene codes for the reverse transcriptase and other enzymes. In addition, HIV-1 contains six genes (vif, vpu, vpr, tat, rev and nef) in its 9kB RNA that contribute to its genetic complexity. Tat and rev are regulatory proteins that accumulate within the nucleus and bind to defined regions of the viral RNA: TAR (transactivation-response elements), found in the LTR; and RRE (rev response elements), found in the env gene, respectively. The tat protein is a potent transcriptional activator of the LTR promoter region and is essential for viral replication in almost all in vitro culture systems. Tat and rev stimulate the transcription of proviral HIV-1 DNA into RNA, promote RNA elongation, enhance the transportation of HIV RNA from the nucleus to the cytoplasm and are essential for translation. Rev is also a nuclear export factor that is important for switching from the early expression of regulatory proteins to the structural proteins that are synthesized later.
Nef may induce downregulation of CD4 (11) and HLA class I molecules from the surface of HIV-1-infected cells, which may represent an important escape mechanism for the virus to evade an attack mediated by cytotoxic CD8+ T cells and to avoid recognition by CD4 T cells. Nef may also interfere with T cell activation by binding to various proteins that are involved in intracellular signal transduction pathways. Vpr seems to be essential for viral replication in non-dividing cells such as macrophages. More recently, vpr was shown to be important for the transport of the viral pre-integration complex to the nucleus and may arrest cells in the G2
phase of the cell cycle. Vpu is important for the virus "budding" process, because mutations in vpu are associated with persistence of the viral particles at the host cell surface. Some recent publications have highlighted a new and important role for vif in supporting viral replication Vif-deficient HIV-1 isolates do not replicate in CD4 T cells, some T cell lines ("non-permissive cells") or in macrophages. Vif-deficient isolates are able to enter a target cell and initiate reverse transcription, but synthesis of proviral DNA remains incomplete
(Fig.)  .
.
.
The HIV cycle
CD4 is a 58 kDa monomeric glycoprotein that can be detected on the cell surface of about 60% of T-lymphocytes, T cell precursors within the bone marrow and thymus and on monocytes and macrophages, eosinophils, dendritic cells and microglial cells of the central nervous system. The identification of the gp120-binding site on the CD4 of CD4 T cells stimulated attempts to use soluble CD4 (sCD4) to neutralize the circulating virus in patients, the aim being the inhibition of viral spread. In contrast, sCD4 was able to induce
conformational changes within the viral envelope that promoted the infection of target cells. The binding of gp120 to CD4 is not only a crucial step for viral entry, but also interferes with intracellular signal transduction pathways and promotes apoptosis in CD4 T cells. In the past couple of years, the idea of blocking CD4 as the primary cellular receptor of HIV has regained interest.
Chemokine receptors as co-receptors for HIV entry
CCR5 is a necessary co-receptor for monocytotropic (M-tropic) HIV-1 isolates. Later, the chemokine receptor CXCR4 (fusin) was described as being the co-receptor used by T celltropic T-tropic) HIV isolates. Monocytotropic (M-tropic) HIV-1 isolates are classically those viruses that are most easily propagated in macrophage cultures, are unable to infect T cell lines (i.e., immortalized T cells), but are able to easily infect primary T cells from peripheral blood samples. Conversely, T cell-tropic HIV-1 isolates have classically been identified as being those that are easily propagated in T cell lines, and grow poorly in macrophages, but
are also able to easily infect primary T cells from peripheral blood samples.
Chemokines (“Chemotactic cytokines") and their receptors have been previously characterized with regard to their role in promoting the migration (chemotaxis) of leukocytes and their pro-inflammatory activity. They are proteins of 68-120 amino acids which depend on the structure of their common cysteine motif, and which may be subdivided into C-X-C (α -chemokines), C-C (ß-chemokines) and C-chemokines. Chemokines typically show a high degree of structural homology to each other and may share the receptors they bind to.
Chemokine receptors belong to the group of receptors with seven transmembrane regions ("7- transmembrane receptors"), which are intracellularly linked to G-proteins. SDF-1 ("stromal cell-derived factor 1") was identified as the natural ligand of CXCR4 and is able to inhibit the entry of T-tropic HIV-1 isolates into activated CD4 T cells. Rantes ("regulated upon activation T cell expressed and secreted"), MIP-1α ("macrophage inhibitory protein") and MIP-1ß represent the natural ligands of CCR5 and are able to inhibit the entry of M-tropic HIV-1 isolates into T cells. T-tropic HIV-1 isolates mainly infect activated peripheral blood CD4 T cells and cell lines and use CXCR4 for entry into the CD4+-positive target cell. Mtropic
isolates are able to infect CD4 T cells, monocytes and macrophages, and depend on the use of CCR5 and CD4 for viral entry. The interaction of gp120 and the cellular receptors is now understood in more detail. gp120 primarily binds to certain epitopes of CD4. Binding to CD4 induces conformational changes in gp120 that promote a more efficient interaction of gp120 with its respective co-receptor. Membrane fusion is dependent on gp120 co-receptor binding. gp41, as the transmembrane part of the envelope glycoprotein gp160, is crucial for the fusion of the viral and the host cell membrane. It was postulated that consequent to the binding of gp120 to CD4, a conformational change is induced in gp41 that allows gp41 to insert its hydrophobic NH2 terminal into the target cell membrane. The identification of crucial amino acid sequences for this process was used to synthesize peptides that bind to gp41 within the domains, are critical for the induction of conformational changes, and that may inhibit membrane fusion.
T20 is the first of several peptides that bind to gp41 and has been tested in clinical trials for suppressing viral replication. Currently, T20 is available as a therapeutic option for selected patients. One disadvantage of T20 is that it must be taken intramuscularly rather than as a pill. Using transfected cell lines, besides CCR5 and CXCR4, other chemokine receptors, such as CCR3, CCR2, CCR8, CCR9, STRL33 ("Bonzo"), Gpr 15 ("Bob"),Gpr 1, APJ and ChemR23, were identified and shown to be used for entry by certain HIV isolates. APJ may represent a relevant co-receptor within the central nervous system. Despite this broad spectrum of potentially available co-receptors, CCR5 and CXCR4 seem to represent the most relevant co-receptors for HIV-1 in vivo. The importance of CCR5 as the predominant coreceptor for M-tropic HIV isolates is underscored by another observation. The majority of individuals with a genetic defect of CCR5 are resistant to infection with HIV-1. In vitro experiments show that lymphocytes derived from these individuals are resistant to HIV-1 infection using M-tropic isolates but not to infection with T-tropic isolates. Lymphocytes
from these individuals do not express CCR5 on their cell surface and genetically have a 32 base pair deletion of the CCR5 gene. Worldwide, a few patients have been identified that have acquired HIV-1 infection despite a homozygous deletion of the CCR5. As expected, all of them were infected with CXCR4-using HIV-1 isolates. In epidemiological studies, the allelic frequency of the CCR5 gene deletion is 10-20% among Caucasians. The frequency of a homozygous individual is about 1% in Caucasians. Studies conducted on African or Asian populations, however, do not find this 32 base pair deletion of the CCR5, suggesting that this mutation arose after the separation of these populations in evolutionary history.
Individuals that are heterozygous for the 32 bp deletion of the CCR5 show a decreased expression of CCR5 on the cell surface and are more frequently encountered within cohorts of long-term non-progressors compared to patients who have a rapid progression of disease. In addition, HIV-infected individuals who are heterozygous for the 32 bp deletion of the CCR5, have a slower progression to AIDS, a better treatment response to HAART and lymphoma incidence is decreased. These data demonstrate that the density of CCR5 on the cell surface is not only a limiting factor for replication of HIV in vitro but also in vivo.
In addition to the 32bp deletion of the CCR5, other genetic polymorphisms, with regard to the chemokine receptors (CCR2) or their promoters (CCR5), have been described. Based on the occurrence of these polymorphisms within defined patient cohorts, they were associated with a more rapid or a more favorable course of disease, depending on the particular polymorphism. In patients who have a rapid progression of disease (rapid drop in CD4 T cell count), virus isolates that use CXCR4 as a predominant co-receptor tend to be frequently isolated from their cells, in comparison to patients with a stable CD4 T cell count. The
expression of co-receptors on CD4+ lymphocytes depends on their activation level. CXCR4 is mainly expressed on naive T cells, whereas CCR5 is present on activated and effector/memory T cells. During the early course of HIV-1 infection, predominantly M-tropic HIV-1 isolates are detected. Interestingly, M-tropic HIV-1 isolates are preferentially transmitted regardless of whether or not the "donor" predominantly harbors T-tropic isolates. At present, it remains unclear whether this "in vivo" preference of M-tropic HIV-1 isolates is determined by selected transportation of M-tropic isolates by sub-mucosally located dendritic cells or whether the local cytokine/chemokine milieu favours the replication of M-tropic viruses. Recent intriguing studies suggest that M-tropic HIV-1 viruses are able to ‘hide' more easily from the immune system by replicating in macrophages, in comparison to T-tropic viruses, thus giving them a survival advantage in the infected individual. The blockade of CCR5, therefore, seems to represent a promising target for therapeutic intervention. In vitro, monoclonal antibodies to CCR5 (2D7 and others) are able to block the entry of CCR5-using HIV isolates into CD4 T cells and macrophages. Small molecule inhibitors of CCR5 have been designed and are currently being tested in clinical trials. In vitro studies, as well as experiments using SCID mice, however, suggest that blockade of CCR5-using isolates may alter their tropism towards increased usage of CXCR4. Small molecule inhibitors such as T22, ALX40-4C or AMD3100 are able to inhibit CXCR4 and are also subject to preclinical and clinical trials. Strategies are currently being developed to modulate expression of chemokine receptors. Intrakines are chemokines that stay within the cytoplasm and are able to capture and bind to their corresponding receptor on its way to the cell surface. "Short interfering RNA" (siRNA) represents a new molecular tool that is able to selectively inactivate target genes. Double-stranded RNA is split by the enzyme dicer-1 into short pieces ("21-23mers"). These oligomers because of complementarity may bind to longer RNA sequences that are then subsequently degraded. This strategy is currently employed in plants and used for its antiviral activity. The use of siRNA against CCR5 can prevent the expression of CCR5 in vitro. Although the therapeutic use of chemokine receptor blockers seems promising, a lot of questions still remain unanswered. Using knockout mice it was demonstrated that the absence of CXCR4 or SDF-1 is associated with severe defects in hematopoiesis and in cerebellar development. Currently, it remains unclear whether the blockade of CXCR4 in postnatal or adult individuals may also affect other organ systems.
Post Fusion Events
Following membrane fusion the virus core "uncoats" into the cytoplasm of the target cell. These "early events" have recently been studied in more detail. HIV can enter into rhesus lymphocytes but replication is stopped before or during early reverse transcription. This intracellular blockade is mediated by a cellular factor, TRIM5α, which is a component of cytoplasmic bodies and whose primary function is not yet known. TRIM5a from various species exhibits differential inhibition on various retroviruses. For example, TRIM5α from rhesus macaques, TRIM5α rh, more profoundly inhibit HIV replication than human TRIM5α, whereas SIV (simian immunodeficiency virus) which naturally infects Old World monkeys, is less susceptible to either form of TRIM5α thus explaining in part the species specificity of HIV for human cells. TRIM5α from human cells or non-human primates is able to inhibit replication of other lentiviruses and represents a novel cellular resistance factor whose definitive biological significance has yet to be fully characterized. It is unclear how exactly TRIM5α blocks reverse transcription and it has been hypothesized that TRIM5a interferes with the incoming virus capsid protein targeting it for ubiquitination and proteolytic degradation.
HIV-1 entry into quiescent T cells is comparable to HIV-1 entry into activated T cells, but synthesis of HIV-1 DNA remains incomplete in quiescent cells. The conversion of viral RNA into proviral DNA, mediated by the viral enzyme reverse transcriptase (RT), occurs in the cytoplasm of the target cell and is a crucial step within the viral replication cycle. Blockade of the RT by the nucleoside inhibitor zidovudine was the first attempt to inhibit viral replication in HIV-1 infected patients. Today, numerous nucleoside, nucleotide and nonnucleoside RT inhibitors are available for clinical use and have broadened the therapeutic arsenal substantially since the mid eighties.
Reverse transcription occurs in multiple steps. After binding of the tRNA primers, synthesis of proviral DNA occurs as a minus-strand polymerization starting at the PBS ("primer binding site") and extending up to the 5' repeat region as a short R/U5 DNA. The next step includes degradation of RNA above the PBS by the viral enzyme RNAase H and a "template switch" of the R/U5 DNA with hybridization of the R sequence at the 3' RNA end. Now the full length polymerization of proviral DNA with degradation of the tRNA is completed.
Reverse transcription results in double-stranded HIV DNA with LTR regions ("long terminal repeats") at each end.
HIV-1 enters into quiescent T cells and reverse transcription may result in the accumulation of proviral, non-integrating HIV-DNA. However, cellular activation is necessary for integration of the proviral HIV DNA into the host cell genome after transportation of the preintegration complex into the nucleus. Cellular activation may occur in vitro after stimulation with antigens or mitogens, in vivo activation of the immune system is observed after antigen contact or vaccination or during an opportunistic infection. In addition, evidence is emerging that HIV-1 gp120 itself may activate the infecting cell to enhance integration. Besides monocytes, macrophages and microglial cells, latently infected quiescent CD4+ T cells that contain non-integrated proviral HIV DNA represent important long-living cellular reservoirs of HIV. Since natural HIV-1 infection is characterized by continuing cycles of viral replication in activated CD4+ T cells, viral latency in these resting CD4+ T cells likely represents an accidental phenomenon and is not likely to be important in the pathogenesis of this disease. This small reservoir of latent provirus in quiescent CD4+ T cells gains importance, however, in individuals who are treated with HAART, since the antivirals are unable to affect non-replicating proviruses and thus the virus will persist in those cells and be replication competent to supply new rounds of infection, if the drugs are stopped. Thus, the existence of this latent reservoir has prevented HAART from entirely eradicating the virus from infected individuals. Until recently it was not clear, why HIV replicates poorly in
quiescent CD4 T cells. The cellular protein Murr1 that plays a role in copper metabolism is able to inhibit HIV replication in unstimulated CD4 T cells. Murr1 was detected in primary resting CD4 T cells and interferes with activation of the transcription factor NFκB by inhibiting the degradation of IκBα . IκBα prevents NFκB from migrating to the nucleus, especially after cytokine stimulation (e.g., TNFα ). Because the HIV LTR region has multiple sites for NFκB, preventing NFκB migration to the nucleus should inhibit HIV replication.
Inhibition of murr-1 by siRNA is associated with HIV replication in quiescent CD4 T cells.
Persistence of HIV in quiescent CD4 T cells and other cellular reservoirs seems one of the main reasons why eradication of HIV is not feasible. If it is ever possible to achieve, a more detailed knowledge of how and when cellular reservoirs of HIV are established and how they may be targeted is of crucial importance for the development of strategies aiming at HIV eradication.
Cellular transcription factors such as NF-κB may also bind to the LTR regions. After stimulation with mitogens or cytokines, NF-κB is translocated into the nucleus where it binds to the HIV-LTR region, thereby initiating transcription of HIV genes. Transcription initially results in the early synthesis of regulatory HIV-1 proteins such as tat or rev. Tat binds to the TAR site ("transactivation response element") at the beginning of the HIV-1 RNA in the nucleus and stimulates transcription and the formation of longer RNA transcripts. Rev
activates the expression of structural and enzymatic genes and inhibits the production of regulatory proteins, therefore promoting the formation of mature viral particles. The proteins coded for by pol and gag form the nucleus of the maturing HIV particle; the gene products coded for by env form the gp120 "spikes" of the viral envelope. The gp120 spikes of the envelope are synthesized as large gp160 precursor molecules and are cleaved by the HIV-1 protease into gp120 and gp41. The gag proteins are also derived from a large 53 kD precursor molecule, from which the HIV-protease cleaves the p24, p17, p9 and p7 gag proteins.
Cleavage of the precursor molecules by the HIV-1 protease is necessary for the generation of infectious viral particles, and therefore the viral protease represents another interesting target for therapeutic blockade. The formation of new viral particles is a stepwise process: a new virus core is formed by HIV-1 RNA, gag proteins and various pol enzymes and moves towards the cell surface. The large precursor molecules are cleaved by the HIV-1 protease, which results in the infectious viral particles budding through the host cell membrane. During the budding process, the virus lipid membranes may incorporate various host cell proteins
and become enriched with certain phospholipids and cholesterol. In contrast to T cells, where budding occurs at the cell surface and virions are released into the extracellular space, the budding process in monocytes and macrophages results in the accumulation of virions within cellular vacuoles.
The replication of retroviruses is prone to error and is characterized by a high spontaneous mutation rate. On average, reverse transcription results in 1-10 errors per genome and per round of replication. Mutations can lead to the formation of replication-incompetent viral species. But, mutations causing drug resistance may also accumulate, which, provided that there is selection pressure under certain antiretroviral drugs and incomplete suppression of viral replication, may become dominant.
In addition, viral replication is dynamic and turns over quickly in infected individuals at an average rate of 109 new virus particles being produced and subsequently cleared per day. Thus, within any individual, because of the extensive virus replication and mutation rates, there exists an accumulation of many closely related virus variants within the ‘population' of viruses, referred to as a viral "quasispecies". The selection pressure on mostly the pre-existing mutations may not only be exerted by certain drugs, but also by components of the immune system, such as neutralizing antibodies or cytotoxic T cells (CTL).
Transmission routes
There are several ways in which someone can become infected with HIV. These transmission routes are well defined. HIV infection can be transmitted through:
1. unprotected sexual intercourse with an infected partner.
1. unprotected sexual intercourse with an infected partner.
2. injection or transfusion of contaminated blood or blood products (infection through artificial insemination, skin grafts and organ transplants is also possible).
3. sharing unsterilized injection equipment that has been previously used by someone who is infected.
4. maternofetal transmission (during pregnancy, at birth and through breast-feeding).
Cause of infection
Acute HIV-1 infection presents in 40 - 90 % of cases as a transient symptomatic illness, associated with high levels of HIV-1 replication and an expansive virus-specific immune response. With 14,000 new cases per day worldwide, it is an important differential diagnosis in cases of fever of unknown origin, maculopapular rash and lymphadenopathy. The diagnosis of acute infection is missed in the majority of cases, as other viral illnesses ("flu") are often assumed to be the cause of the symptoms, and there are no HIV-1-specific antibodies detectable at this early stage of infection. The diagnosis therefore requires a high degree of clinical suspicion, based on clinical symptoms and history of exposure, in addition to specific laboratory tests (detection of HIV-1 RNA or p24 antigen and negative HIV-1 antibodies) confirming the diagnosis. An accurate early diagnosis of acute HIV-1 infection is important, as patients may benefit from therapy at this early stage of infection (see below), and infection of sexual partners can be prevented.
Immunological and virological events during acute HIV-1 infection
During acute HIV-1 infection, the virus is replicating extensively in the absence of any detectable adaptive immune response, reaching levels of up to 100 million copies of HIV-1 RNA/ml. It is during this initial cycle of viral replication that important pathogenic processes are thought to occur. These include the seeding of virus to a range of tissue reservoirs and the destruction of HIV-1-specific CD4+ T lymphocytes. Several factors can influence viral replication during acute infection and the establishment of a viral set point. These include the
fitness of the infecting virus, host genetic factors and host immune responses. While antibodies against HIV-1 with neutralizing capacities are rarely detectable during primary HIV-1 infection, a number of studies have demonstrated a crucial role of HIV-1-specific cellular immune responses for the initial control of viral replication during this stage of infection. A massive, oligoclonal expansion of CD8+ T cell responses has been described during acute HIV-1 infection, and the appearance of HIV-1-specific CD8+ T cells has been
temporally associated with the initial decline of viraemia. These CD8+ T cells have the ability to eliminate HIV-1-infected cells directly by MHC class I-restricted cytolysis or indirectly by producing cytokines, chemokines or other soluble factors, thus curtailing the generation of new viral progeny. The biological relevance of HIV-1-specific cytotoxic T cells (CTL) in acute HIV-1 infection was highlighted in recent in-vivo studies demonstrating a dramatic rise of SIV viraemia and an accelerated clinical disease progress in macaques after the artificial depletion of CD8+ T cells. Additional evidence for the antiviral pressure of HIV-1-specific CTLs during primary HIV-1 infection has been provided by the rapid selection of viral species with CTL epitope mutations that were detected within a few weeks after HIV-1 and SIV infection in humans and rhesus macaques, respectively.
During acute HIV-1 infection, the number of CD4+ T cells decline, occasionally to levels that allow the development of opportunistic infections at that time. Even though the CD4+ T cell count rebounds with the resolution of primary infection, it rarely returns to baseline levels in the absence of antiretroviral therapy. In addition to the decline in CD4+ T cell counts, qualitative impairments of CD4+ T cell function are perhaps the most characteristic abnormalities detected in HIV-1 infection. The impairment of HIV-1-specific CD4+ T cell
During acute HIV-1 infection, the number of CD4+ T cells decline, occasionally to levels that allow the development of opportunistic infections at that time. Even though the CD4+ T cell count rebounds with the resolution of primary infection, it rarely returns to baseline levels in the absence of antiretroviral therapy. In addition to the decline in CD4+ T cell counts, qualitative impairments of CD4+ T cell function are perhaps the most characteristic abnormalities detected in HIV-1 infection. The impairment of HIV-1-specific CD4+ T cell
function occurs very early in acute infection, potentially due to the preferential infection of virus-specific CD4+ T cells by the virus. This is followed by a functional impairment of CD4+ T cell responses to other recall antigens, as well as a reduced responsiveness to novel antigens.
In addition to host immune responses, host genetic factors play an important role in both susceptibility and resistance to HIV-1 infection and speed of disease progression following infection. The most important of these is a deletion in the major co-receptor for entry of HIV- 1 into CD4+ T cells, a chemokine receptor called CCR5. Homozygotes for this 32 base pair deletion (CCR5delt32) do not express the receptor at the cell-surface and can only be infected with HIV strains that are able to use other co-receptors, such as CXCR4. Thus, although CCR5delta32 homozygotic individuals show a significant degree of resistance to HIV-1 infection, a number of cases of infection with CXCR4-using HIV-1 strains have been described. Heterozygotes for the deletion exhibit significant lower viral setpoints and slower progression to AIDS. In addition to mutations in the chemokine receptor genes, a number of HLA class I alleles have been described to be associated with both, lower viral setpoints and slower disease progression, including HLA-B27 and -B57. Recent studies demonstrated that individuals expressing HLA-B57 presented significantly less frequently with symptomatic acute HIV-1 infection and exhibited a better control of viral replication following acute infection. These data demonstrate that host genetic factors can influence the clinical manifestations of acute HIV-1 infection and have an important impact on subsequent viral setpoints and the speed of disease progression.
Signs and symptoms
After an incubation period of a few days to a few weeks after exposure to HIV, most infected individuals present with an acute flu-like illness. Acute HIV-1 infection is a very heterogeneous syndrome and individuals presenting with more severe symptoms during acute infection and a longer duration of the acute infection syndrome tend to progress more rapid to AIDS. The clinical symptoms of acute HIV-1 infection were first described in 1985 as an illness resembling infectious mononucleosis. The most common symptoms are fever,
maculopapular rash, oral ulcers, lymphadenopathy, arthralgia, pharyngitis, malaise, weight loss, aseptic meningitis and myalgia. In a recently published study by Hecht et al., fever (80 %) and malaise (68 %) had the highest sensitivity for clinical diagnosis of acute HIV-1 infection, whereas loss of weight (86 %) and oral ulcers (85 %) had the highest specificity. In this study, the symptoms of fever and rash (especially in combination), followed by oral ulcers and pharyngitis had the highest positive predictive value for diagnosis of acute HIV-1 infection. In another study by Daar et al., fever, rash, myalgia, arthralgia and night sweats were the best predictors for acute HIV-1 infection. The symptomatic phase of acute HIV-1 infection lasts between 7 - 10 days, and rarely longer than 14 days. The nonspecific nature of the symptoms poses a great challenge to the clinician and underlines the importance of a detailed history of exposure.
Diagnosis
The diagnosis of acute HIV-1 infection is based on the detection of HIV-1 replication in the absence of HIV-1 antibodies, as these are not yet present at this early stage of infection. Different tests are available for diagnosis of acute HIV-1 infection. The most sensitive tests are based on detection of plasma HIV-1 RNA. During acute HIV-1 infection, there is frequently a marked decrease of CD4+ cell count, which later increases again, but usually does not normalize to the initial levels. In contrast, the CD8+ cell count rises initially, which
may result in a CD4+/CD8+ ratio of < 1. Infectious mononucleosis is the most important differential diagnosis. Hepatitis, Influenza, toxoplasmosis, syphilis and side effects of medications may also be considered. In summary, the most important step in the diagnosis of acute HIV-1 infection is to include it in the differential diagnosis. The clinical suspicion of an acute HIV-1 infection then merely requires performance of an HIV-1 antibody test confirmed by using an ELISA kit with a different type of antigen crating from the first kit (viral lysate, recombinant antigens, or synthetic peptides) and possibly repeated testing of HIV-1 viral load.
Treatment
The goal of antiretroviral therapy during acute HIV-1 infection is to reduce the number of infected cells, preserve HIV-1-specific immune responses and possibly lower the viral set point in the long term. Several studies in recent years have shown that treatment of acute HIV-1 infection allows long-term viral suppression, leads to preservation and even increase of HIV-1-specific T helper cell responses and allows for the conservation of a very homogeneous virus population. First pilot studies in patients who were treated during acute HIV-1 infection and subsequently went through structured treatment interruptions show that the HIV-1-specific immune response could be boosted in these patients. Most patients were subsequently able to discontinue therapy and experienced at least temporal control of viral replication, with viral set points remaining below 5,000 copies/ml for more than 3 years in some patients. However, in a large number of individuals (>50%) in this study, as well as in other studies assessing viral control following treated primary infection, viral load rebounded during longer follow-up, requiring the initiation of therapy. The long-term clinical benefit of early initiation of therapy has not been demonstrated yet. It is also not known how long the
period between acute infection and initiation of therapy can be without losing immunological, virological and clinical benefit. In view of all these unanswered questions, patients with acute HIV-1 infection should be treated in controlled clinical trials. If this is not possible, the option of standard first-line treatment should be offered and discussed. Usually, treatment continues for at least a year, followed by structured treatment interruptions within the framework of controlled studies or other immunotherapeutic intervention. It is important during counselling to clearly indicate the lack of definitive data on clinical benefit and to address the risks of antiretroviral therapy and treatment interruptions, including drug toxicity, development of resistance, acute retroviral syndrome during viral rebound and HIV-1 transmission and superinfection during treatment interruptions.
D. Influenza virus
True influenza is an acute infectious disease caused by a member of the orthomyxovirus family: Influenza virus A, B or, to a much lesser extent, Influenza virus C. However, the term 'flu' is often used for any febrile respiratory illness with systemic symptoms that may be caused by a myriad of viral or rarely bacterial agents as well as influenza. Influenza outbreaks usually occur in the winter in temperate climates. Influenza is commonly seen in local outbreaks or epidemics throughout the world. Epidemics may appear at any time but are usually concentrated in months of high humidity. They occur explosively with little or no warning. The number of people affected can vary from a few hundred to hundreds of thousands. Epidemics may be short-lived, lasting days or weeks but larger epidemics may last for months. Although influenza is a mild disease in most individuals, it is life threatening in elderly or debilitated individuals. Epidemics are responsible for large losses in productivity.
Influenza virus particles are highly pleiomorphic (variable), mostly spherical/ovoid, 80-120nm diameter, but many forms occur, including long filamentous particles (up to 2000nm long x 80-120nm diameter). Different strains of virus vary in their tendency to form filaments- this property maps to the matrix protein. The outer surface of the particle consists of a lipid envelope from which project prominent glycoprotein spikes of two types:
• Haemagglutinin (HA), a 135Å envelope spike is a trimeric protein called the haemagglutinin (HA) of which there are 13 major antigenic types. The haemagglutinin functions during attachment of the virus particle to the cell membrane, and can combine with specific receptors on a variety of cells including red blood cells.
• Neuraminidase (NA), a 60Å tetramer 
The matrix protein lines the inner side of the envelope. This electron micrograph shows several influenza virus particles (at a magnification of about 284,000x). The surface projections are molecules of haemagglutinin and neuraminidase The genome segments are packaged into the core. The RNP (RNA + nucleoprotein, N) is in a helical form with the 3-polymerase polypeptides associated with each segment.


Influenza viruses are taken into the cell by endocytosis. In the low pH environment of the endosome, RNP is released from MP1, and the viral lipoprotein envelope fuses with the lipidbilayer of the vesicle, releasing viral RNP into the cell cytoplasm, from where it is transported into the nucleus. New viral proteins are translated from transcribed messenger RNA (mRNA). New viral RNA is encased in the capsid protein, and together with new matrix protein is then transported to sites at the cell surface where envelope haemagglutinin and neuraminadase components have been incorporated into the cell membrane. Progeny virions are formed and released by budding  Influenza virus showing RNP complex
Influenza virus showing RNP complex
Influenza virus showing RNP complex
Pathogenesis and disease
The virus spreads from person to person via small particle aerosols (less than 10μm) which can get into respiratory tract. It can also survive for a short time on surfaces and can be spread by this route if the virus is introduced into the nasal mucosa before it loses infectivity. The incubation period is short, about 18 to 72 hrs. Virus concentration in nasal and tracheal secretions remains high for 24 to 48 hrs after symptoms start and may last longer in children.
Titers are usually high and so there are enough infectious particles in a small droplet to start a new infection. Influenza virus infects the epithelial cells of the respiratory tract. The cells die, in part due to the direct effects of the virus on the cell, and also possibly due to the effects of interferon. Cell death at later times may also result from the actions of cytotoxic T-cells. As a result, the efficiency of ciliary clearance is reduced, leading to impaired function of the mucus elevator; thus there is reduced clearance of infectious agents from the respiratory tract.
Recovery
Interferon may play a role by decreasing virus production. Many of the symptoms of uncomplicated influenza (muscle aches, fatigue, fever) are associated with the efficient induction of interferon. The cell-mediated immune response is important in viral clearance. The antibody response is usually not significant until after virus has been cleared. Repair of the respiratory epithelium begins rapidly, but may take some time to complete. A humoral antibody response is the main source of protection. IgG and IgA are important in protection against reinfection. Antibody to the HA protein is most important since this can neutralise the virus and prevent the virus initiating the infection. Neutralisation frequently involves blocking of the binding of the virus to host cells and may work at other steps involved in the entry and uncoating of the virus. Antibody to the NA protein has some protective effect since it seems to slow the spread of the virus. IgG persists longer than IgA and so plays a more important role in long-term immunity.
Diagnosis
Firm diagnosis is by means of virus isolation and serology. The virus can be isolated from the nose or a throat swab. This is used to infect cells in culture (or eggs). Haemadsorption may be used to detect infected cells. Polymerase chain reaction (PCR) tests are being developed to detect viral RNA. Recently, rapid tests that can be used in a physician's office have been approved. Provisional diagnosis is often made clinically, based on knowledge of a current outbreak of influenza combined with appropriate clinical symptoms.
Vaccines
A new vaccine is formulated annually with the types and strains of influenza predicted to be the major problems for that year (predictions are based on worldwide monitoring of influenza). The vaccine is multivalent and the current one is against two strains of influenza A and one of influenza B. The vaccine given to adults at present is an inactivated preparation of egg-grown virus. Only certain formulations of the vaccine are approved for young children. Previously, a subunit vaccine was recommended. In 2003, a live, attenuated (much less pathogenic than wild-type virus) vaccine was approved for use. It is given nasally and should provide mucosal, humoral and cell-mediated immunity. In this vaccine, the vaccine virus is a cold-adapted strain, which can grow in the upper respiratory tract where it is cooler, but grows poorly in the lower respiratory tract.
Chemotherapy
Rimantadine and amantadine block virus entry across the endosome and also interfere with virus release. They are good prophylactic agents for influenza A, but there are some problems in taking them on a long term basis. They may be given as protective agents during an outbreak, especially to those at severe risk and key personnel. They may also be given at the time of vaccination for a few weeks, until the humoral response has time to develop.
Vaccines in prevention of viral infections
A vaccine is an antigenic preparation used to produce active immunity to a disease, in order to prevent or ameliorate the effects of infection by any natural or 'wild' strain of the organism. The term derives from vaccinia, the infectious viral agent of cowpox ("vaca" means cow in Latin), which, when administered to humans, provided them protection against smallpox. The process of distributing and administrating vaccines is referred to as vaccination.
Types of vaccines
Vaccines may be living, weakened strains of viruses or bacteria, which intentionally give rise to inapparent-to-trivial infections. Vaccines may also be killed or inactivated organisms or purified products derived from them. There are three types of traditional vaccines:
a) Inactivated - these are previously virulent micro-organisms that have been killed with chemicals or heat. Examples are vaccines against flu, cholera, bubonic plague, and hepatitisA. Most such vaccines may have incomplete or short-lived immune responses and are likely to require booster shots.
b) Live, attenuated - these are live micro-organisms that have been cultivated under conditions which disable their virulent properties. They typically provoke more durable immunological responses and are the preferred type for healthy adults. Examples include yellow fever, measles, rubella, and mumps.The live tuberculosis vaccine is not the contagious TB strain, but a related strain called "BCG" is used.
c) Toxoids - these are inactivated toxic compounds from micro-organisms in cases where these (rather than the micro-organism itself) causes illness. Examples of toxoid-based vaccines include tetanus and diphtheria.
A number of innovative vaccines are in development and also in use:
Conjugate - certain bacteria have polysaccharide outer coats that are poorly immunogenic. By linking these outer coats to proteins (e.g. toxins), the immune system can be led to recognize the polysaccharide as if it were a protein antigen.
Subunit - rather than introducing a whole inactivated or attenuated micro-organism to an immune system, a fragment of it can create an immune response.
Recombinant vector - by combining the physiology of one micro-organism and the DNA of the other, immunity can be created against diseases that have complex infection processes.
DNA vaccination - in recent years a new type of vaccine, created from an infectious agent's DNA called DNA vaccination, has been developed. It works by insertion (and expression, triggering immune system recognition) into human or animal cells, of viral or bacterial DNA. These cells then develop immunity against an infectious agent, without the effects other parts of a weakened agent's DNA might have.
Vaccines have contributed to the eradication of smallpox, one of the most contagious and deadly diseases known to man. Other diseases such as rubella, polio, measles, mumps, chickenpox, and typhoid are nowhere near as common as they were just a hundred years ago.
Smallpox vaccine
Smallpox is a serious, contagious, and sometimes fatal infectious disease. There is no specific treatment for smallpox disease, and the only prevention is vaccination. The name smallpox is derived from the Latin word for “spotted” and refers to the raised bumps that appear on the face and body of an infected person. There are two clinical forms of smallpox. Variola major is the severe and most common form of smallpox, with a more extensive rash and higher fever. Variola minor is a less common presentation of smallpox, and a much less severe disease, with death rates historically of 1% or less. The smallpox vaccine was the first successful vaccine ever to be developed and remains the only effective preventive treatment for the deadly smallpox disease. Edward Jenner first perfected it in 1796. Smallpox outbreaks have occurred from time to time for thousands of years, but the disease is now eradicated after a successful worldwide vaccination program. Generally, direct and fairly prolonged face-to-face contact is required to spread smallpox from one person to another. Smallpox also can be spread through direct contact with infected bodily fluids or contaminated objects such as bedding or clothing. Rarely, smallpox has been spread by virus carried in the air in enclosed settings such as buildings, buses, and trains. Humans are the only natural hosts of variola. Smallpox is not known to be transmitted by insects or animals. A person with smallpox is sometimes contagious with onset of fever (prodrome phase), but the person becomes most contagious with the onset of rash. At this stage the infected person is usually very sick and not able to move around in the community. The infected person is contagious until the last smallpox scab falls off. The smallpox vaccine helps the body develop immunity to smallpox. The vaccine is made from a virus called vaccinia which is a “pox”-type virus related to smallpox. The smallpox vaccine contains the “live” vaccinia virus—not dead virus like many other vaccines. For that reason, the vaccination site must be cared for carefully to prevent the virus from spreading. Smallpox vaccination provides high-level immunity for 3 to 5 years and decreasing immunity thereafter. If a person is vaccinated again later, immunity lasts even longer. Historically, the vaccine has been effective in preventing smallpox infection in 95% of those vaccinated. In addition, the vaccine was proven to prevent or substantially lessen infection when given within a few days of exposure. It is important to note, however, that at the time when the smallpox vaccine was used to eradicate the disease, testing was not as advanced or precise as it is today, so there may still be things to learn about the vaccine and its effectiveness and length of protection.
Polio vaccine
Polio, caused by an enterovirus, is usually spread via the faecal-oral route; i.e., the virus is transmitted from the stool of an infected person to the mouth of another person from contaminated hands or objects such as utensils. Some cases may be spread directly via an oral-oral route. The incubation period of polio is commonly 6-20 days, with a range of 3-35 days. Surprisingly, 95% of all individuals infected with polio have no apparent symptoms. Another 4%-8% of infected individuals have symptoms of a minor, non-specific nature, such as sore throat and fever, nausea, vomiting, and other common symptoms of any viral illness.
About 1%-2% of infected individuals develop nonparalytic aseptic (viral) meningitis, with temporary stiffness of the neck, back, and/or legs. Less than 2% of all polio infections result in the classic "flaccid paralysis," where the patient is left with permanent weakness or paralysis of legs, arms, or both. Patients infected with the poliovirus can pass the virus on for 7-10 days before the onset of disease. In addition, they can continue to shed the virus in their stool for 3-6 weeks. There is no "cure" for polio. Persons infected with polio need supportive therapy, such as bed rest and fluids. Standard precautions should be taken to avoid passing on the virus through any contamination from the patient's stool.
Two polio vaccines are used throughout the world to combat polio. The first was invented by Jonas Salk and first tested in 1952 and announced to the world by Salk on April 12, 1955. It consists of an injected dose of killed polio virus. Thereafter, Albert Sabin produced an oral polio vaccine using live but weakened (attenuated) virus. Human trials of Sabin's vaccine began in 1957 and it was licensed in 1962. When the live-virus Sabin vaccine was developed, it spread in popularity for several reasons.
First, it can 'infect' other, non-vaccinated individual and confer some immunity, referred as herd-immunity, in them.
Second, because the oral vaccine acts in the gut, it confers local immunity and reduces the spread of the wild virus. The injected vaccine, acting through the bloodstream, immunises the individual but does not reduce his ability to spread the wild virus.
Third, the live-virus vaccine is cheaper than the killed-virus vaccine.
Finally, the oral vaccine is easier to administer than the injected vaccine, so patients are more likely to complete the vaccination series and attain full immunity.
Though Salk's vaccine had reduced the incidence of polio to a tiny fraction of what it was in the early 1950s, Sabin's vaccine was considered superior for these reasons and became the standard treatment. The major disadvantage of the live-virus vaccine is that it can itself cause sporadic cases of polio either from the vaccine
Finally, the oral vaccine is easier to administer than the injected vaccine, so patients are more likely to complete the vaccination series and attain full immunity.
Though Salk's vaccine had reduced the incidence of polio to a tiny fraction of what it was in the early 1950s, Sabin's vaccine was considered superior for these reasons and became the standard treatment. The major disadvantage of the live-virus vaccine is that it can itself cause sporadic cases of polio either from the vaccine
itself or from the circulation of the vaccine. There is neither risk from the killed-virus vaccine. The two vaccines have eliminated polio from most of the countries in the world and reduced early cases from hundreds of thousands per year to only 1238 worldwide in 2006.
HIV vaccine
An HIV vaccine is a hypothetical vaccine against HIV, the etiological agent of AIDS. As there is no known cure for AIDS, the search for a vaccine has become part of the struggle against the disease. The urgency of the search for a vaccine against HIV stems from the AIDS-related death toll of over 25 million people since 1981. Indeed, in 2002, AIDS became the primary cause of mortality due to an infectious agent in Africa (UNAIDS, 2004). Highly active antiretroviral therapy (HAART) has been highly beneficial to many HIV-infected individuals since its introduction in 1996, when the protease inhibitor-based HAART initially
became available. Many HIV-infected individuals have experienced remarkable improvements in their general health and quality of life, which has led to the plummeting of HIV-associated morbidity and mortality. Still, for some patients - and in many clinical cohorts this may be more than fifty percent of patients - HAART achieves far less than optimal results. This is due to a variety of reasons such as medication intolerance/side effects,
prior ineffective antiretroviral therapy and infection with a drug-resistant strain of HIV.
However, non-adherence and non-persistence with antiretroviral therapy is the major reason most individuals fail to get any benefit from and develop resistance to HAART. The reasons for non-adherence and non-persistence with HAART are varied and overlapping. Major psychosocial issues, such as poor access to medical care, inadequate social supports, psychiatric disease and drug abuse, contribute to non-adherence. However, the complexity of these HAART regimens, whether due to pill number, dosing frequency, meal restrictions or other issues, undeniably have a weighty impact along with side effects that create intentional
non-adherence. HAART allows the stabilisation of the patient’s symptoms and viraemia, but they do not cure the patient of HIV, nor of the symptoms of AIDS, and high levels of HIV-1, often HAART resistant, return once treatment is stopped. Moreover, it would take more than the lifetime of an individual to be cleared from HIV infection using HAART. Antivirals are also too expensive for developing countries, which have the highest rates of HIV-infection.
Only a vaccine will be able to halt the pandemic. This would possibly cost less, thus being affordable for developing countries, and would not require daily treatment. However, after over 20 years of research, HIV remains a difficult target for a vaccine.
Problems with HIV vaccine research
The classical vaccination approaches that have been successful in the control of various viral diseases by priming the adaptive immunity to recognise the viral envelope proteins have failed in the case of HIV-1, as the epitopes of the viral envelope are too variable.
Furthermore, the functionally important epitopes of the gp120 protein are masked by glycosylation, trimerisation and receptor-induced conformational changes making it difficult to block with neutralising antibodies. There are two key issues that protect HIV from vaccines:
Firstly, selective pressure quickly neutralised early promise, the falling off of immune response is called anergy. HIV responds rapidly to these pressures. From human isolates it has been discovered that HIV currently has three groups of clades, M, N and O. Nine clades have been identified in M (A-G) but fewer in the others. The earliest vaccine were based on the LAI clade, which was discovered to be rare in human infections.
The second problem with HIV is its attack on the immune system itself which means that, to date, no effective cell-mediated immune response has been determined. Although there have been more than 50 million infections in Africa since 1981, there have been few vaccine trials in the continent that has been the most severely afflicted by the pandemic. HIV-1 vaccines need to be developed for testing in Africa, as it is in this context, with the risk of infection being the greatest, that a preventative vaccine can be proved to be effective. The remarkable genetic diversity of HIV-1 strains in Africa seems is a major hurdle in the development of a
broadly effective vaccine. All known HIV-1 subtypes and an array of intersubtype recombinant viruses, often in complex mosaic forms, are found throughout the continent, however, the major HIV-1 subtypes accounting for most infections in Africa are subtype C in southern Africa, subtypes A and D in eastern Africa. Therefore, CTL and antibody responses raised by any vaccine need to be broad enough to cover this variability. The typical animal model for vaccine research is the monkey, often the macaque. The monkeys can be infected
with SIV or the chimeric SHIV for research purposes. However, the well-proven route of trying to induce neutralizing antibodies by vaccination has stalled because of the great difficulty in stimulating antibodies that neutralise heterologous primary HIV isolates. Some vaccines based on the virus envelope have protected chimpanzees, or macaques, from homologous virus challenge, but in clinical trials, individuals that were immunised with similar constructs became infected after later exposure to HIV-1. The human body can defend
itself against HIV, as work with monoclonal antibodies (MAb) has proven. That certain individuals can be asymptomatic for decades after infection is encouraging. Enormous effort has been put into understanding how HIV works, it has produced a number of approaches to vaccination, none of which have been effective. Methods attempted include recombinant proteins, synthetic peptides, recombinant viral vectors, recombinant bacterial vectors, recombinant particles, DNA vaccines to induce production of a specific antigen, and whole killed and live-attenuated HIV, though these latter two have not progressed into clinical trials in uninfected individuals due to an unfavorable benefit/risk ratio. The role of broadly neutralising antibodies (NAb) is under investigation, although earlier results were discouraging. vaccines are used to investigate the HIV glycoproteins. Attacks on particular parts of the RNA code of the virus have shown some promise, such as those against the nef gene which regulates viral replication. The burden of HIV-related disease and transmission in Africa demands that enhanced efforts be made to treat large numbers of infected people with
antiretroviral drugs, despite the cost and the emergence of resistant strains, while effective preventative vaccines are developed. Also, access to care and the development of simplified monitoring schemes for positive and negative treatment effects, as well as effective planning on how to obtain and distribute HIV vaccines needs to be addressed. This will be an important step if a large scale phase III HIV-vaccine trial is to be effective (WHO-UNAIDS, 2001), as this may bypass what we saw with antiretroviral therapy, that drugs were initially widely used in only developed countries.
Clinical trials to date
Seventeen vaccine candidates are in in phase I trials and four in phase I/II. There is only one in phase III. Most initial approaches focused on the HIV envelope protein. At least thirteen different gp120 and gp160 envelope candidates have been evaluated, in the US. Most research focused on gp120 rather than gp41/gp160, as the latter are generally more difficult to produce and did not initially offer any clear advantage over gp120 forms. Overall, they have been safe and immunogenic in diverse populations, have induced neutralizing antibody in nearly 100% recipients, but rarely induced CD8+ cytotoxic T lymphocytes (CTL).
Mammalian derived envelope preparations have been better inducers of neutralizing antibody than candidates produced in yeast and bacteria. Although the vaccination process involved many repeated "booster" injections, it was very difficult to induce and maintain the high antigp120 antibody titres necessary to have any hope of neutralising an HIV exposure. The availability of several recombinant canarypox vectors has provided interesting results that may prove to be generalizable to other viral vectors. Increasing the complexity of the
canarypox vectors by inclusion of more genes/epitopes has increased the percent of volunteers that have detectable CTL to a greater extent than did increasing the dose of the viral vector. Importantly, CTLs from volunteers were able to kill peripheral blood mononuclear cells infected with primary isolates of HIV, suggesting that induced CTLs could have biological significance. In addition, cells from at least some volunteers were able to kill cells infected with HIV from other clades, though the pattern of recognition was not uniform among volunteers. As canarypox is the first candidate HIV vaccine that has induced crossclade
functional CTL responses, other strategies that have progressed to phase I trials in uninfected persons include peptides, lipopeptides, DNA, an attenuated Salmonella vector, lipopeptides, p24, etc. Specifically, candidate vaccines that induce one or more of the following are being sought: broadly neutralizing antibody against HIV primary isolates; cytotoxic T cell responses in a vast majority of recipients; strong mucosal immune responses.
Experimental vaccine has been developed by Merck to stimulate HIV-specific cellular immunity, which prompts the body to produce T cells that kill HIV-infected cells. In previous smaller trials, this vaccine was found to be safe and to induce cellular immune responses against HIV in more than half of volunteers. The Merck vaccine contains a weakened adenovirus that serves as a carrier for three subtype B HIV genes. Subtype B is the most prevalent HIV subtype in the regions of the study sites. Adenoviruses are among the main causes of upper respiratory tract ailments such as the common cold. Because the vaccine contains only three HIV genes housed in a weakened adenovirus, study participants cannot become infected with HIV or get a respiratory infection from the vaccine. Novel approaches, including modified vaccinia Ankara (MVA), adeno-associated virus, Venezuelan Equine Encephalitis (VEE) replicons, and codon-optimised DNA have proven to be strong inducers of CTL in macaque models, and have provided at least partial protection in some models. Most of these approaches are, or will soon, enter clinical studies.
Suggested Readings
1. Flint, S. J., V. R. Racaniello., L. W. Enquist., R. M. Krug., A. M. Skalka. 2000. Principles of Virology: molecular biology, pathogenesis, and control. ASM Press, Washington, DC.
2. Dimmock, N. J., Dimmock, N., Easten, A.J. (Andrew), Leppard, K. 2006. Introduction to modern virology. Sixth edition. Blackwell Pub., New York.
3. Suttle, C. A. Viruses in the sea. 2005. Nature, (vol.437) Pg356-361.
4. Black, J.G. 1996. Microbiology. Principles and Applications. Third Edition. Prentice Hall. Upper Saddle River, New Jersey. pp. 274-280.
5. Hunt, Richard. Microbiology and Immunology on-line. University of South Carolina.
My name is hoover, my 18 years old daughter, Tricia was diagnosed of herpes 3 years ago. ever since then,we have been going from one hospital to the other. We tried all sorts of pills but all efforts to get rid of the virus was futile. The blisters kept on reappearing after some months. My daughter was making use of Acyclovir tablets 200mg. 2 tablets every 6hours and fusitin cream 15grams. and H5 POT. Permanganate with water to be applied 2x a day but all still show no result. So I was on the internet some months back, to sought for any other means of saving my only child. just then, i came across a comment on dr imoloa herbal treatment and decided to give it a try. i contacted the him and he prepared some herbs and sent it to me together with guidelines on how to use the herbs through DHL courier service. my daughter used it as directed dr imoloa and in less than 14days, my daughter regained her health.. You should contact Dr imoloa today directly on his email address for any kind of health challenge; lupus disease, mouth ulcer, mouth cancer, body pain, fever, hepatitis A.B.C., syphilis, diarrhea, HIV/AIDS, Huntington's Disease, back acne, Chronic renal failure, addison disease, Chronic Pain, Crohn's Disease, Cystic Fibrosis, Fibromyalgia, Inflammatory Bowel Disease, fungal nail disease, Lyme Disease, Celia disease, Lymphoma, Major Depression, Malignant Melanoma, Mania, Melorheostosis, Meniere's Disease, Mucopolysaccharidosis , Multiple Sclerosis, Muscular Dystrophy, Rheumatoid Arthritis, Alzheimer's Disease, parkison disease, vaginal cancer, epilepsy, Anxiety Disorders, Autoimmune Disease, Back Pain, Back Sprain, Bipolar Disorder, Brain Tumour, Malignant, Bruxism, Bulimia, Cervical Disk Disease, cardiovascular disease, Neoplasms, chronic respiratory disease, mental and behavioural disorder, Cystic Fibrosis, Hypertension, Diabetes, asthma, Inflammatory autoimmune-mediated arthritis. chronic kidney disease, inflammatory joint disease, impotence, feta alcohol spectrum, Dysthymic Disorder, Eczema, tuberculosis, Chronic Fatigue Syndrome, constipation, inflammatory bowel disease. and many more; contact him on email- drimolaherbalmademedicine@gmail.com./ also on whatssap-+2347081986098.
ReplyDelete